Définition / fréquence
La macrodactylie est définie, selon Barsky, par une augmentation de volume de tous les éléments d’un ou de plusieurs doigts.
Il s’agit d’une anomalie rare représentant 0,9% des anomalies congénitales au membre supérieur selon Flatt. Le premier cas a été décrit en 1824 (Klein).
Classification
Plusieurs classifications ont été décrites. Nous en retiendrons deux :
- Flatt et Kelikian différencient les macrodactylies selon le tissu pathologique impliqué:
- Type I : la lipofibromatose. Le doigt est infiltré par un tissu fibro-graisseux.
- Type II : la neurofibromatose. Elle correspond au développement d’un neurofibrome plexiforme.
- Type III : l’hyperostose digitale. Il s’agit d’une anomalie très rare, présente à la naissance, et comportant le développement de masses ostéochondrales épiphysaires. L’anomalie est souvent bilatérale.
- Type IV : l’hémihypertrophie
- Temtamy et McKusik (1978) différentient les augmentations de volume du doigt isolées de celles associées à un syndrome :
- Les macrodactylies non syndromiques sont divisées en :
- Macrodactylies vraies : elles correspondent au type I de Flatt.
- Pseudo macrodactylies en rapport avec une malformation vasculaire ou lymphatique augmentant le volume du doigt. On peut y ajouter l’hyperostose digitale (type III de Flatt)
- Les macrodactylies syndromiques accompagnent des syndromes suivants :la neurofibromatose, le gigantisme partiel et l’hémihypertrophie, le syndrome de Protée, la maladie d’Ollier, le syndrome de Maffucci, le syndrome de Klippel-Trenaunay-Weber et le lymphoedème congénital (Milroy)
- Les macrodactylies non syndromiques sont divisées en :
Les syndromes comportant une augmentation de volume des doigts ou des mains
- Le syndrome de Klippel-Trenaunay-Weber associe hypertrophie d’un membre, naevi cutanés et dilatations veineuses.
- Le syndrome de McCune-Albright comprend la triade dysplasie fibreuse polyostotique souvent unilatérale, taches cutanées café-au-lait, et puberté précoce. Sa cause est inconnue. La cavité médullaire contient un tissu fibreux peu ossifié avec un aspect en verre dépoli sur la radiographie. Sa transformation maligne est possible en ostéo ou fibrosarcome.
- L’enchondromatose multiple (Maladie d’Ollier) correspond au développement intra osseux de chondromes. Ceux-ci peuvent entraîner des troubles de croissance. Elle touche souvent un hémicorps. L’association à des angiomes réalise le syndrome de Mafucci.
- Le syndrome de Protée (Wiedemann) a été décrit en 1983 et porte ce nom en référence à la mythologie grecque. Proteus, divinité marine et fils de Poseidon, avait le don de prophétie et la capacité de se transformer à volonté pour échapper aux mortels qui lui demandaient des prophéties. Il associe gigantisme partiel des mains et/ou des pieds, naevi pigmentés, hémi hypertrophie, tumeurs sous cutanées (hamartomes), anomalies squelettiques (protubérances osseuses) et faciales, croissance accélérée et anomalies viscérales.
- La neurofibromatose (Type III de Flatt). La neurofibromatose de type I (Maladie de Recklinghausen) est une maladie autosomique dominante avec des mutations spontanées. La fréquence est de 1/3000 naissances. L’atteinte des mains n’est pas au premier plan mais lorsqu’elle est présente, elle comporte le développement d’un neurofibrome plexiforme et de masses ostéochondrales épiphysaires. Parmi les autres éléments du syndrome, on retiendra : plus de 5 taches café au lait, des tumeurs cutanées ou nerveuses, des anomalies squelettiques (pseudarthroses congénitales de jambe, scolioses dystrophiques dans 30% des cas), un gliome du chiasma et parfois un retard mental. Les risques de cancer sont importants. Du point de vue anatomo pathologique, il s’agit d’un « neurofibromes plexiformes » associant fibrose épi et périneurale sur des nerfs sinueux et une absence d’infiltration graisseuse. Un suivi génétique et neuro pédiatrique est indispensable.
- L’hémi hypertrophie (Type IV de Flatt) (Voir ci-dessus)
Les vraies macrodactylies (Type I de Flatt)
Elles correspondent à l’infiltration fibro lipomateuse d’un ou de plusieurs doigts. Elles sont rares et doivent être distinguées de ces autres causes. Son incidence est faible. Le premier cas a été décrit en 1824.
L’étiologie est inconnue. La théorie la plus communément admise est une anomalie localisée des récepteurs aux facteurs de croissance (Ochi). Il n’y a habituellement pas d’ATCD familial ou d’anomalie chromosomique rapportée.
Deux éléments caractérisent ce type de macrodactylie :
- L’infiltration graisseuse de l’ensemble du doigt prédominant en palmaire
- L’accélération de la croissance squelettique dans toutes les directions
Deux évolutions sont possibles selon sa vitesse de croissance:
- Forme statique: la macrodactylie est présente à la naissance et croit de façon proportionnelle par rapport au reste de la main.
- Forme progressive: plus fréquente, peut ne pas être présente à la naissance, croit rapidement vers 2 ans et atteint son pic de croissance en fin de puberté. Cette forme est souvent sévère avec croissance disproportionnée en longueur et largeur.
Il est difficile à la naissance de faire la différence entre ces deux formes.

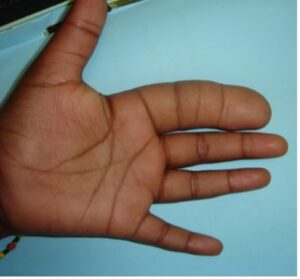
Du point de vue clinique, l’atteinte peut être uni ou pluridigitale. Le deuxième rayon est le plus souvent atteint, suivi du troisième et du premier. Plusieurs doigts sont plus fréquemment atteints (70%) qu’un seul. La face palmaire est plus souvent atteinte entraînant une hyper extension interphalangienne et un élargissement des phalanges. L’atteinte du pouce l’entraîne en extension et abduction. Une syndactylie est parfois associée (10%). Dans les formes sévères, l’extension de l’infiltration atteint à la main et l’avant-bras. Lors des atteintes des 2ème et 3ème rayons, le doigt est incurvé vers le coté ulnaire. La mobilité est habituellement conservée lorsque l’enfant est jeune.
Les radiographies montrent une croissance accélérée avec avancement de l’âge osseux. Il peut y avoir un élargissement de l’espace inter métacarpien. A l’âge adulte apparaissent des ostéophytes.
Il existe une masse molle, non douloureuse, pouvant siéger sur la paume de la main, la première commissure et/ou le 1/3 distal avant-bras. Le territoire de distribution est habituellement celui du médian. Celui-ci peut présenter un aspect en sablier au niveau du canal carpien entraînant parfois un syndrome canalaire. L’infiltration peut gagner les nerfs digitaux. La sensibilité discriminative reste bonne chez l’enfant.
Les formes à orientation nerveuse associent une macrodactylie et une infiltration graisseuse du nerf innervant la zone impliquée dans la macrodactylie appelée lipofibrome. 40 à 60% des lipofibromes comportent une macrodactylie.
L’échographie retrouve un élargissement fusiforme du nerf avec prolifération graisseuse dissociant les fascicules et un rétrécissement dans le canal carpien.
L’IRM retrouve une augmentation de volume du nerf avec des fascicules dissociés par un infiltrat graisseux. Le nerf a souvent un aspect sinueux.
Traitement
Plusieurs traitements ont été proposés : résection de la peau et du tissu sous cutané, résection nerveuse, épiphysiodèse, ostéotomie, arthrodèse et amputation. Ces procédures peuvent être combinées entre elles et vont dépendre de l’âge du patient et du type et de l’importance de l’atteinte.
—-
en cours